What is phylogeny?
Phylogeny is the study of species evolutionary relationships. Evolutionary relationships can be estimated by the comparison of characteristics and other information about the organism (1). With information about each organism scientists are able to construct phylogenetic trees.
What is a phylogenetic tree?
Professional phylogenetic trees are created with an assortment of information about an organism. This information can range from the morphology to specific DNA sequences. Using this information, scientists will look for potential trees that can explain the evolutionary path that each species followed. Phylogenetic trees can be created with many different sources of information; however, the evolution of sequencing technologies has made DNA or protein sequences the primary source of information (2). Sequences can be aligned with multiple different types of software, such as ClustalOmega and Mega. Differences in the sequence can be detected, and a tree can be formed through neighbor joining, maximum likelihood, and maximum parsimony methods.
Building a phylogenetic tree for ALDH2
1) Collect homologous sequences.
2) Align sequences using ClustalOmega or MegaX.
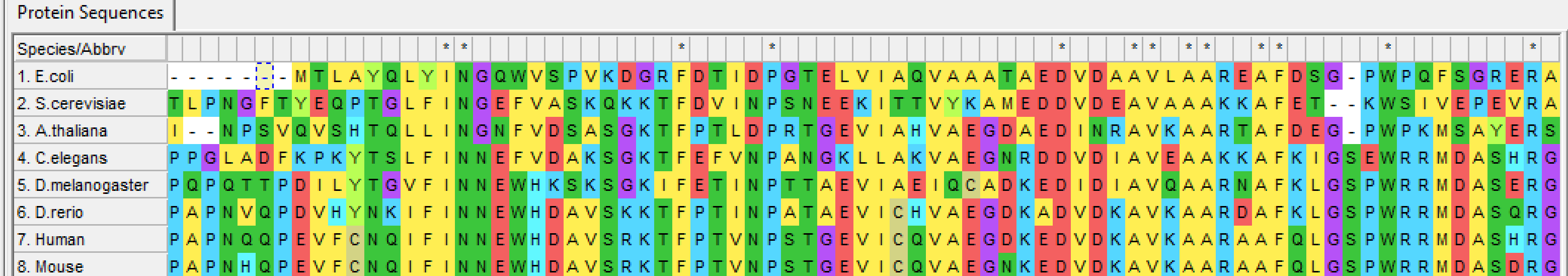
Figure 1: ALDH2 alignment using Mega
3) Build trees using MegaX.
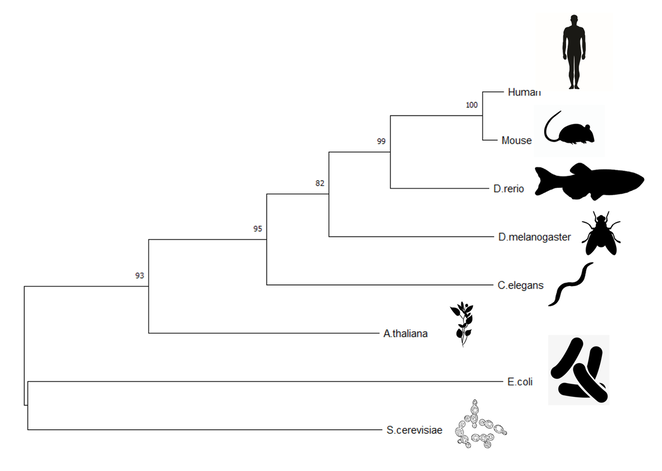
Figure 2: Maximum likelihood tree constructed using MegaX
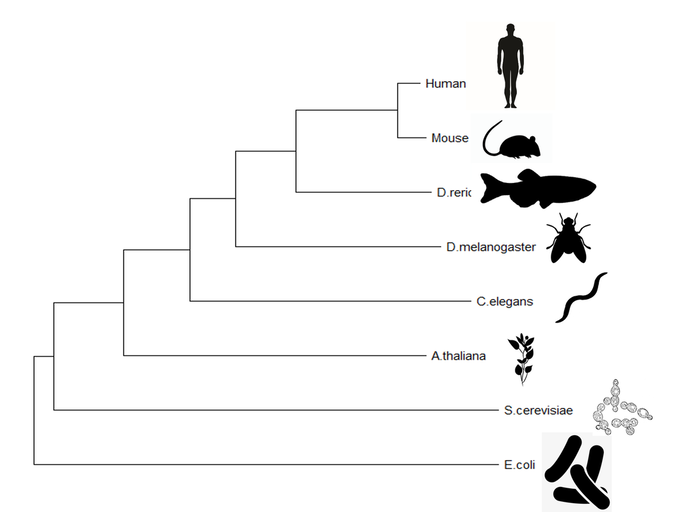
Figure 3: ALDH2 neighbor joining tree made using MegaX
Discussion
Both the maximum likelihood and neighbor joining tree predict an evolutionary model for the ALDH2 homologs between species. The trees display similar evolutionary pathways, but differ in some of the nodes of the tree. These phylogenies demonstrate the relationship of the mammals, where the mouse model is a very close relative of the human protein. As expected the unicellular organisms are the most distant relatives.
